
Introduction
As I already revealed in the last post, I intend to have several projects with Density Functional Theory on this blog. I already have a simple project on GitHub, about a ‘quantum dot’1 with volumetric visualization of orbitals with VTK.
I thought that exposing some theory in a separate post would be nice for further references, so without further ado, here it is.
Links
On this blog
The How to solve a quantum many body problem post is relevant, it contains topic related things like Born–Oppenheimer approximation and variational principle. The post on Hartree-Fock is also relevant, up to the Hartree term discussion. Actually, all of it is relevant but I’ll detail that later. The section about basis sets is also important. I used plane-waves for the DFT code, but it could be changed to use something else, like Gaussians or wavelets. Also the last post, Solving Poisson Equation contains related information.
Some pdf documents
I’m not going to give a lot of links here, one can find a lot of them on the net, but a couple shouldn’t hurt. Here is the Nobel Lecture of W. Kohn2. A very nice read, but if you want something more detailed, here is another one: A bird’s-eye view of density-functional theory by Klaus Capelle3. Another one would be The ABC of DFT by Kieron Burke & friends4. Since I mentioned Hartree-Fock, here is a paper with a comparison: Density Functional Theory versus the Hartree Fock Method: Comparative Assessment5. Here is a link from where you can download some lecture notes: Electron Density functional theory by Roi Baer6 Last but not least, I should mention a paper I already mentioned last post: New Algebraic Formulation of Density Functional Calculation Sohrab Ismail-Beigi, T. A. Arias7.
Video lectures
As for the pdf documents case, one can find plenty of video lectures on the net, this is a very big subject. Nevertheless, besides the video about the practical aspects I already pointed to in the last post, I’ll put here this introductory one:
Besides this, I’ll point to a recent set of video lectures from CECAM: Teaching the Theory in Density Functional Theory8. They are more detailed, so if the above video is easy for you, you might want to see some of those.
Some theory
Kohn Sham equations
As in the Hartree-Fock case, I’ll briefly present here some sort of justification (but it’s hand waving, really, you should check out the real derivations). The theory requires some variational calculus (as in the Hartree-Fock case) and it would be too much to type here in LaTex, so for the details one could follow the above links.
In the Hartree-Fock post I presented an equation containing the Fock term:

Here is written a little differently, but it’s the same thing. The second term contains the nuclear potential and perhaps some other external potential, the third one has the sum moved after the integral sign then the sum is done to obtain the density. This is relatively easy to solve but it has big issues, being a mean field theory equation. This equation neglects electron correlations and the Pauli exclusion principle is not obeyed. For molecules it gets energies too high, bindings too weak and distances between atoms too big. Electrons lower the total energy by avoiding each other and make the bindings stronger.
The Hartree-Fock equation tries to solve the issue (along with eliminating the self-interaction that appears due of the Fock term) by adding the exchange term. But that neglects the electrons correlations.
It turns out that one can try to take into account correlations, too, by using an additional exchange-correlation potential term. By adding such exchange-correlation term, one gets the Kohn-Sham equations:
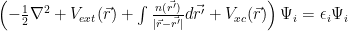
The resemblance with the Hartree-Fock equations should be obvious. For Hartree-Fock, only exchange is taken into account and the potential is a non-local one.
For DFT, the potential is obtained from the exchange-correlation energy functional: ![V_{xc}(\vec{r}) = \frac{\delta E_{xc}[n]}{\delta n(\vec{r})}](https://s0.wp.com/latex.php?latex=V_%7Bxc%7D%28%5Cvec%7Br%7D%29+%3D+%5Cfrac%7B%5Cdelta+E_%7Bxc%7D%5Bn%5D%7D%7B%5Cdelta+n%28%5Cvec%7Br%7D%29%7D+&bg=ffffff&fg=000&s=0&c=20201002)
The density is the usual:
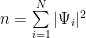
This makes the Kohn-Sham equations, similarly with the Hartree-Fock ones, self-consistent equations. One could solve them by guessing a density for the electrons, use it to solve the Kohn-Sham equations, get the new density out of the solutions and so on, repeating until convergence. For details, please see the Hartree-Fock posts.
Local Density Aproximation
Unfortunately the correlation-exchange functional is not known exactly, so approximations are used. For the purpose of this blog, the local-density approximation is good enough, so I’ll use only that one:
![E_{xc}[n]=\int n(\vec{r})\epsilon_{xc}(n)d\vec{r}](https://s0.wp.com/latex.php?latex=E_%7Bxc%7D%5Bn%5D%3D%5Cint+n%28%5Cvec%7Br%7D%29%5Cepsilon_%7Bxc%7D%28n%29d%5Cvec%7Br%7D&bg=ffffff&fg=000&s=0&c=20201002)
It’s worth mentioning that there are other approximations, like the generalized gradient approximation, where besides the local density also the gradient of density is used, or the meta-GGA, where not only the fist derivative but also the second derivative is used. There are also hybrid approaches using the Hartree-Fock exchange.
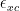
One more thing I want to mention here is that one could use LSDA instead of LDA (S comes from ‘spin’). The main idea is to work with two densities, 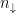

Solving
There are various ways one could try solving the Kohn-Sham equations. One way would be to attack the problem in real space. It’s easy to implement the code like that (but not so easy to do it efficiently) and it’s easy to understand. If you want to go that way, you should check this paper: Real-space mesh techniques in density functional theory by Thomas Beck9.
Another way would be to use Gaussian orbitals. For molecules calculations it would be a very good choice. One could take the already implemented Hartree-Fock program and change it to be also able to perform DFT. That should not be hard at all, in comparison with the difficulty of implementing the Hartree-Fock one. I considered doing that, but because I want to use DFT in more projects than for molecules and because I don’t want to make that project more complex than it is, I gave up the idea.
Another approach would be to start a program from scratch, writing the code in the way described by Tomas Arias7 in the lectures I pointed to in the last post and use the Gaussian orbitals instead of the plane waves basis. The relevant code for the Gaussian orbitals could be taken from the Hartree-Fock program.
Anyway, I decided to use plane waves basis. For a crystal it is a better choice because of the periodic boundary conditions. Working with the reciprocal lattice is a natural choice.
Conclusion
Here I presented briefly some DFT theory for further reference. I might edit the post to add some things in the future, but it’s unlikely that I’ll do substantial additions. The links expose enough theory for understanding the programs.
- ‘Quantum Dot’ The first DFT project on GitHub ↩
- Nobel Lecture W. Kohn ↩
- A bird’s-eye view of density-functional theory by Klaus Capelle ↩
- The ABC of DFT by Kieron Burke & friends ↩
- Density Functional Theory versus the Hartree Fock Method: Comparative Assessment M.Ya. Amusia, A.Z. Msezane, V.R. Shaginyan ↩
- Electron Density functional theory by Roi Baer ↩
- New Algebraic Formulation of Density Functional Calculation Sohrab Ismail-Beigi, T. A. Arias ↩ ↩
- Teaching the Theory in Density Functional Theory CECAM video lectures ↩
- Real-space mesh techniques in density functional theory by Thomas Beck ↩
