
Introduction
Again, I have a new project on GitHub. It’s not so new, it was working already last year (the APW part) but I didn’t have the patience to write a description for it until now. Actually, there are two new projects on GitHub, related, but this post is about the Augmented Plane Waves1 one. Some things I’ll point out in this post are relevant to the KKR project, too. The description for that project will follow in another post.
Program in action
Here is the program running:
At that time, I didn’t have LAPW working, so it shows only the APW results.
Theory
The muffin tin
The periodicity of the lattice suggests dealing with Bloch waves as basis functions and an approach that uses plane waves could be very useful, as it could be seen for the DFT for a quantum dot project. Unfortunately, although that approach has clear advantages, there are still issues. For example, near nucleus the field is very strong and the big kinetic energy of electrons translates in a lot of plane waves needed for the accurate wavefunction description. This can be solved by using a pseudopotential. A pseudopotential is also used in this project and the next one (KKR), but, as I’ll point out later, you can also do computations with a ‘full potential’ in the end, using the muffin tin approach, this is just one more simplification for the beginning. The muffin tin approximation takes into account the strong field close to the nuclei and the fact that ‘far away’ from them the potential varies much slowly, to approximate it with a field that is spherically symmetric inside spheres centered on the atomic nuclei and constant outside (a constant that can be taken to be zero, since you can choose the energy reference point – it’s the differences in energy that matter, not the absolute energy).
As a note, as you dive deeper in the theory for APW and KKR, you find that many simplifying assumptions can be improved, for example the spherical symmetry assumption can be relaxed, or the shape of the muffin potential can take some other form, not necessarily spheres, or the spheres can overlap and so on. By default, this project and the next one assume touching spheres, although for example you may try some smaller spheres with some pseudopotential for Al that could work ok, because for aluminum the nearly free electrons approximation is already relatively good.
Augmented plane waves
Using the above simplifying assumptions, we can try to build up some better basis functions for the solution. The idea is to use the solutions of the Schrodinger equation inside the sphere and plane waves outside the spheres. Since we assumed spherical symmetry we can solve inside the sphere the radial Schrodinger equation only (1D, so it’s relatively easy to solve) as in the DFTAtom project case and with spherical harmonics we have the full solutions. To obtain a basis function we just join a linear combination of solutions of the Schrodinger equation with a plane wave. For that, the plane wave is expanded in spherical harmonics and by requiring equality on the boundary of the sphere we fix the expansion coefficients and so we obtain the augmented plane wave. In the process we limit the maximum azimuthal quantum number to some max value (currently 8 in the project). Then we just have to solve the general eigenvalue equation to get the coefficients for expressing the actual wavefunction in terms of the augmented plane waves. This sounds easy enough, but there is a problem, the solutions of the Schrodinger equation are energy dependent. This makes the generalized eigenvalue equation quite difficult to solve, because it’s non-linear. The program just scans the entire energy interval using a small step, locating changes in sign of the determinant or values that are close to zero (a change of sign might not exist if there is a degeneracy, for example, or if energies are very close and the code steps over a couple). As a detail, in this description a lot of things are not apparent, for example the derivative discontinuity at the sphere boundary in the augmented plane wave is not exactly nice, to see how that is handled you’ll have to check the referred books or papers.
Linearization: LAPW
The energy dependence makes obtaining the solutions very difficult, you can notice that already by comparing the speed of running APW versus LAPW in the project. The non-linearity would make implementing ‘full potential’ (as in a self consistent program using DFT) very difficult. An idea to fix this would be to just pick an energy at half of the energy interval of interest and do computations with that. Unfortunately, the solutions to the Schrodinger equation vary strongly with energy, so this would not give good results. A better approach is to take energy dependence into account by considering also the energy derivative of the solution to the Schrodinger equation. Using that, it is assumed that the solution to the Schrodinger equation has a linear dependence on the energy, around the fixed energy we chose. Again we do the expansion of the plane wave with spherical harmonics, but we match on the sphere boundary not only the value, but also the energy derivative. I won’t give more details here, I’ll end up this by telling that we have to solve the generalized eigenvalue equation, but this time it’s linear, so it can be solved relatively easy.
A possible next step
I might do that in the future, but for now, it’s only a suggestion for extension: Instead of using a pseudopotential, one could use a ‘full potential’ approach in a self-consistent way, that is, use DFT to find the solutions starting from a reasonable electron density, use those solutions to compute a new electron density and so on until convergence is obtained. Probably a lot of the code from DFTAtom could be used. I’ll provide some links that will detail the method more (including code).
Books
Again the Computational Physics book by Jos Thijssen2 must be mentioned. Both APW and LAPW are described in chapter 6. And again, another book is Electronic Structure, Basic Theory and Practical Methods by Richard M Martin3. APW is in chapter XVI, LAPW is in chapter XVII.
By the way, never assume that the formulae in books are correct, for example 6.44a from Computational Physics has a 

Articles and so on
I won’t point many articles for APW, instead of that I’ll point out a couple on LAPW, because it’s more important:
This one basically describes the LAPW as implemented in the project: Use of energy derivative of the radial solution in an augmented plane wave method: application to copper by D D Koelling and G O Arbman4.
This one is worth looking into for both LAPW and also LMTO which is related with the next topic: Linear methods in band theory by O. Krogh Andersen5.
Those are lecture notes from Rutgers University: Application of DFT to Crystals by Kristjan Haule6. Although the ideas are correct, I must warn you that they are full of tiny mistakes, I checked a little more carefully the LAPW part and I noticed several: for example, the versor of the sum is not the same as the sum of versors, they have a 
The code
Some details
Again, the code is here, on GitHub1.
The classes related with APW are in the APW namespace. The BandStructureBasis class is very similar with the BandStructure class you can find in the Tight Binding project or the Empirical Pseudopotential one. I simply got it from the Empirical Pseudopotential project, along with the SymmetryPoint and SymmetryPoints classes and changed it a bit. The actual computation happens in the derived class, BandStructure, with the help of the Numerov classes (for solving the Schrodinger equation, check out the DFTAtom project for another example that does that) and Secular which contains the computation of matrices elements and the determinant of the secular equation.
The LAPW namespace contains classes related with LAPW, but the computation also uses classes from the APW namespace. You’ll find in there the Integral class (which I got from the DFTAtom project) and the Hamiltonian matrix, which deals with matrix elements computations and solving the generalized eigenvalue problem. As for APW, the computation happens in the Compute function of the BandStructure class, but from the LAPW namespace this time.
Both APW and LAPW implementations use a non linear grid for solving the Schrodinger equation.
As a funny detail about how I implemented it, I added the LAPW code in a hurry, trying to have it working as fast as possible. Of course I made a mistake (actually several, but this one was the one that delayed me), passing the index k (an integer) instead of the actual 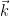
Libraries used
For the user interface, I used wxWidgets9. For the chart, I used VTK10 and for the matrix stuff, I used as usually for the projects for this blog, Eigen11.
Conclusion
If you have suggestions or you find bugs, please let me know. I might sometime implement a full potential LAPW, but for now – except the next post, for which the project is already done – I’ll switch to other topics, I have already four projects dealing with band structure computation in the repository, so for a while those should be enough.
- APW Project on GitHub ↩ ↩
- Computational Physics book by Jos Thijssen ↩
- Electronic Structure, Basic Theory and Practical Methods by Richard M Martin ↩
- Use of energy derivative of the radial solution in an augmented plane wave method: application to copper by D D Koelling and G O Arbman ↩
- Linear methods in band theory by O. Krogh Andersen ↩ ↩
- Application of DFT to Crystals by Kristjan Haule, Rutgers University ↩
- APW The APW project for the above lecture ↩
- LAPW The LAPW project for the Rutgers lecture ↩
- wxWidgets Cross-platform GUI library ↩
- VTK, The Visualization Toolkit The scientific (and not only) visualization library ↩
- Eigen The matrix library ↩
