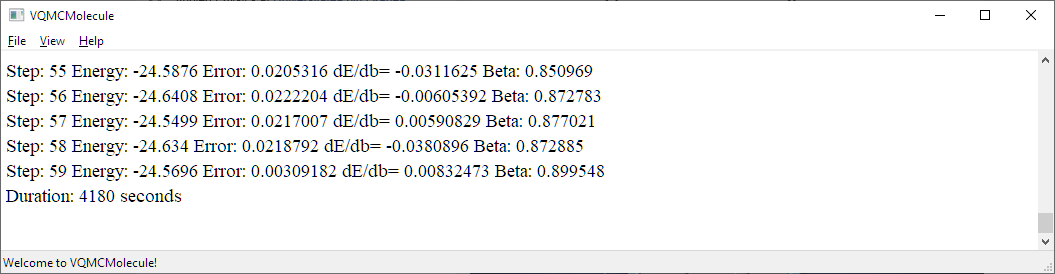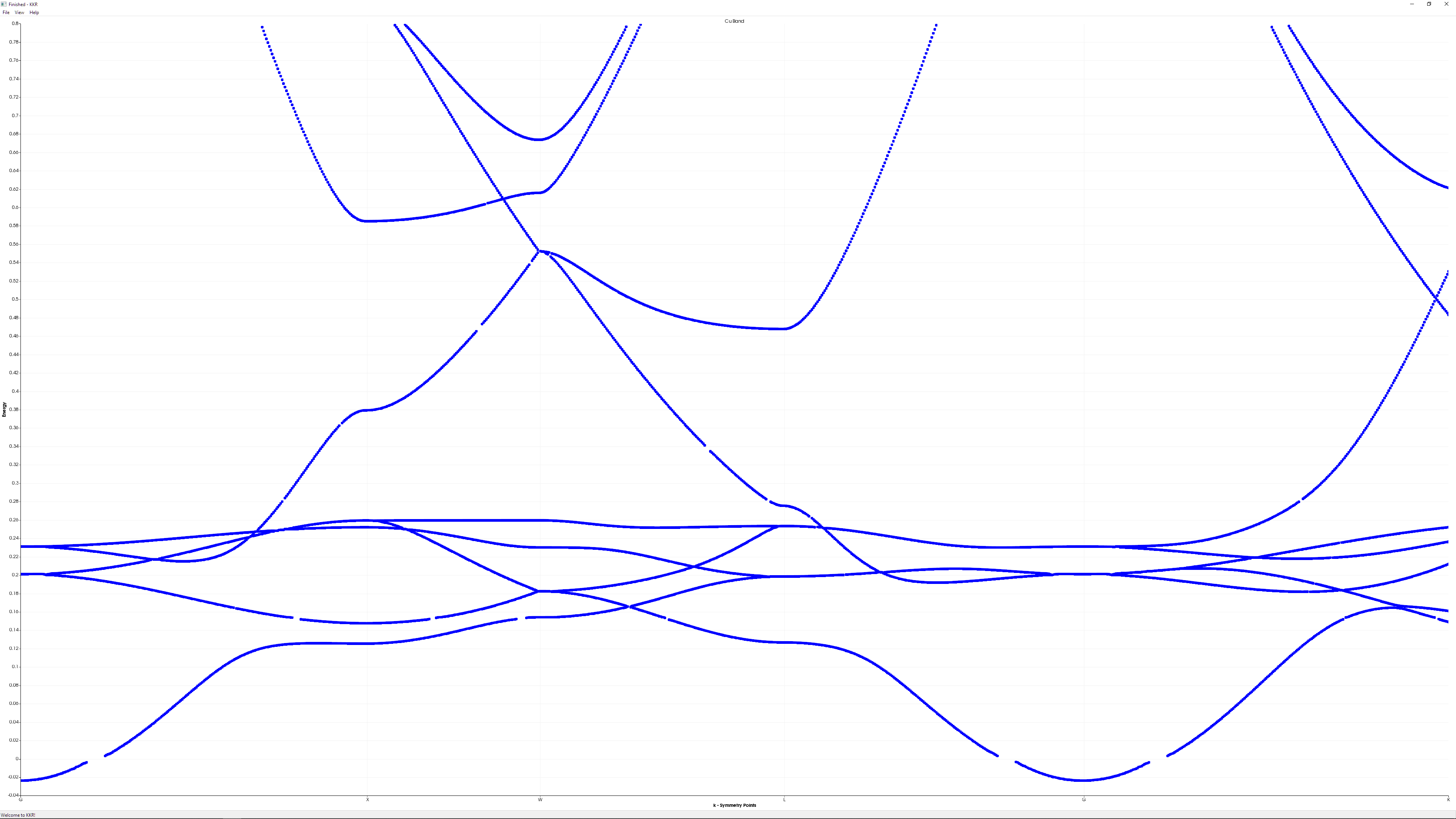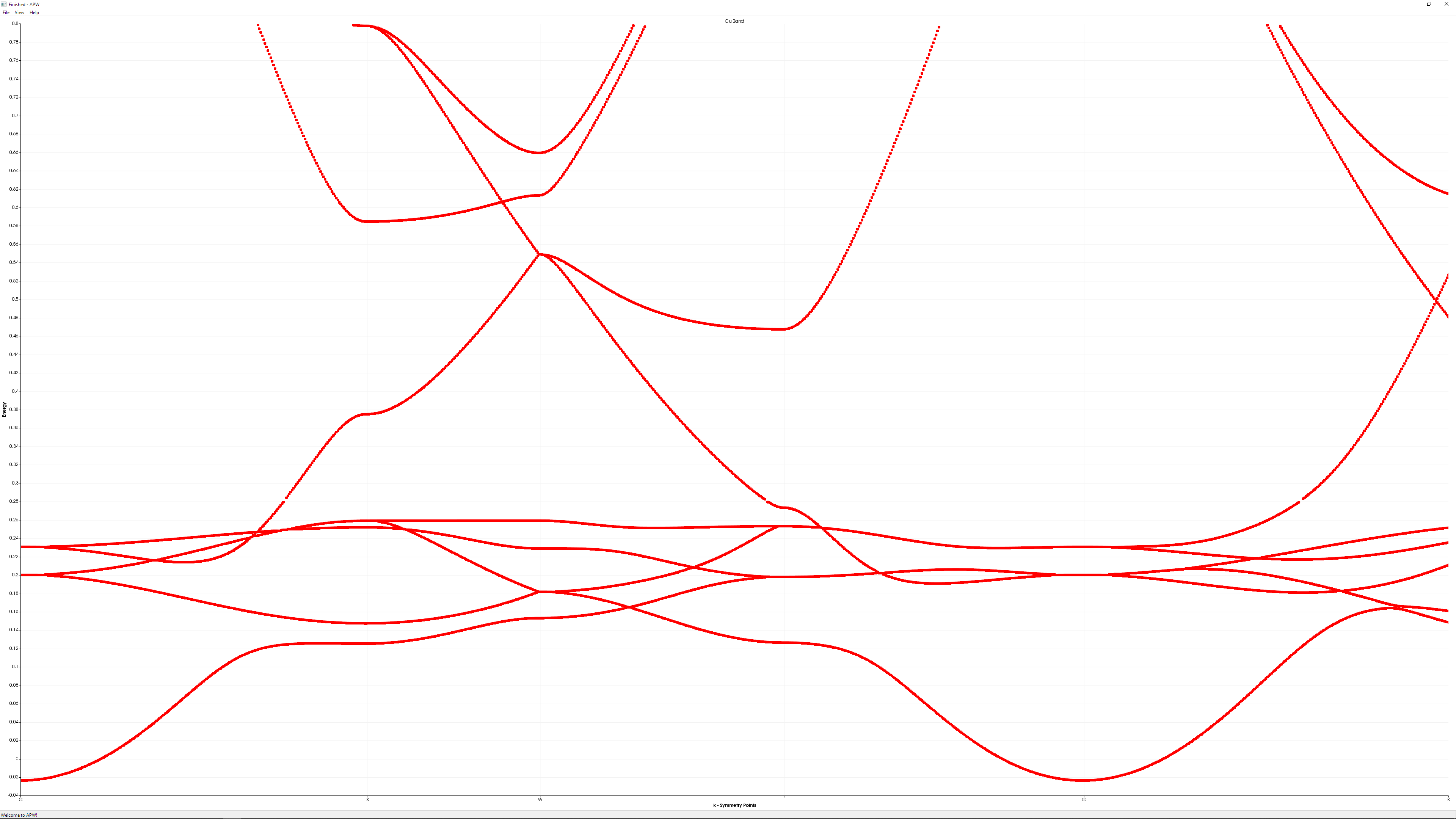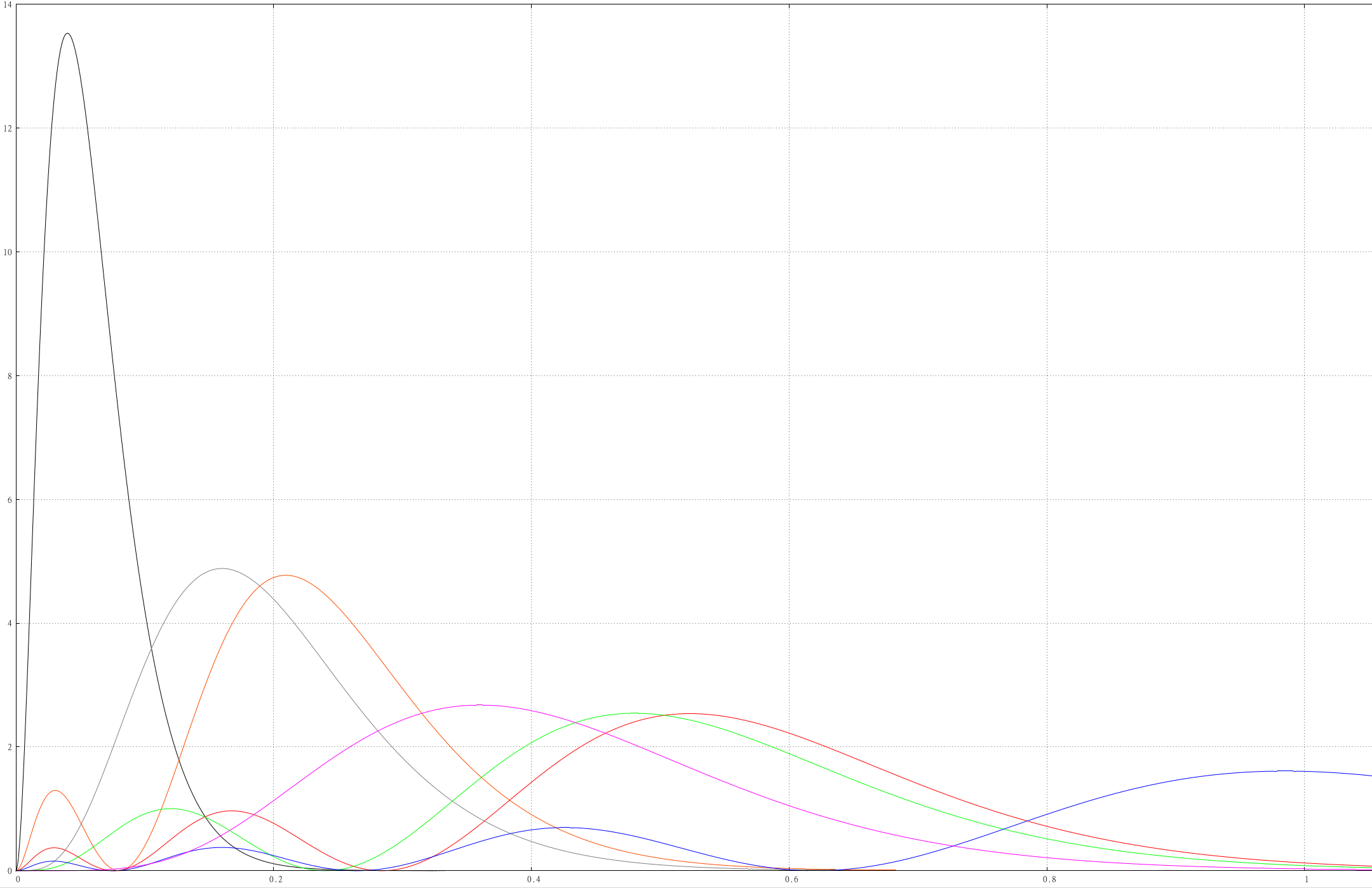
Car-Parrinello Quantum Molecular Dynamics
Introduction I have another repository on GitHub. I decided to have one where I’ll put python code for computational physics issues that are simpler / less complete than the code for the C++ projects. I’ll put there both jupyter notebooks and python scripts. At this moment there are only two of them, hopefully I’ll add … Read more